The identification of peptides that can stimulate Cytotoxic T Lymphocytes (CTLs) is one of the major challenges in subunit vaccines design. Most of existing epitope prediction methods are based on identification of MHC binding peptides. It is not necessary the all MHC binders can act as T cell epitopes. Thus, there is a need to develop a highly accurate prediction method for CTL epitopes instead of MHC binders. The use of artificial neural network and support vector machine on the recent and high quality CTL epitopes and non-epitopes data is explored as a means to meet these challenges.
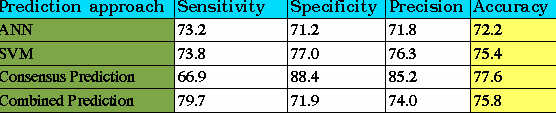
Here, machine learning techniques SVM and ANN have been used to develop a CTL epitope prediction method. First, an ANN based method has been developed for classify the large dataset of CTL epitopes (1137) and non-epitopes (1134). With the implementation of feed-forward backpropagation type of network 72.2% accuracy is achieved. Further, an SVM model was generated by applying SVM_LIGHT on same dataset. The optimized SVM classified the data with 75.2% accuracy, which is better in comparison to ANN. The accuracy of methods was estimated through leave one out cross-validation (LOOCV) at a cutoff score where sensitivity and specificity are nearly equal. In the last, both prediction methods were combined for utilizing their complete potential in classifying the data. The consensus and combined prediction resulted in improvement of specificity and sensitivity respectively along with accuracy. The best accuracy obtained by consensus and combined prediction approaches are 77.6% and 75.8% respectively, which is significantly higher as compared to the individual methods.The overall architecture of the prediction method is shown in figure below.

The overall architecture of CTLpred.
The method is divided in three parts
1) Data extraction and Preprocessing.
2) Training and Testing of methods
3) Consensus and Combined Prediction Approaches.
Where E means epitopes, NE means Non-epitopes, LOOCV means Leave One Out Cross Validation, SNNS means Stuttgart Neural Networks System, SVM means Support Vector Machine, PE means Predicted Epitopes,PNE means Predicted Non-epitopes,Cnp means Consensus Prediction and Cbp means Combined Prediction.
 Artificial Neural Network-: The artificial neural networks are crude electronic model based on the structure of brain.A network can consist of a few to a few billion neurons connected in an array of different methods. ANNs attempt to model these biological structures both in architecture and operation.The basic computational element (model neuron) of neural network is often called a node or unit. It receives input from some other units, or perhaps from an external source. Each input has an associated weight w, which can be modified. The unit computes some function f of the weighted sum of its inputs.
Artificial Neural Network-: The artificial neural networks are crude electronic model based on the structure of brain.A network can consist of a few to a few billion neurons connected in an array of different methods. ANNs attempt to model these biological structures both in architecture and operation.The basic computational element (model neuron) of neural network is often called a node or unit. It receives input from some other units, or perhaps from an external source. Each input has an associated weight w, which can be modified. The unit computes some function f of the weighted sum of its inputs.
Its output, in turn, can serve as input to other units.The weighted sum is called the net input to unit i, often written neti.
Note that wij refers to the weight from unit j to unit i (not the other way around). The function f is the unit's activation function. In the simplest case, f is the identity function, and the unit's output is just its net input. This is called a linear unit..

Basic Architecture-: The basic architecture of artificial neural network is shown through figure below

There are many types of networks ranging from simple networks(Perceptrons) to complex self -orgnising networks(Kohonen networks).Similarly, there are many different kinds of learning rules used by neural networks, the common being the delta rule. The delta rule is often utilized by the most common class of ANNs called 'backpropagational neural networks' (BPNNs). Backpropagation is an abbreviation for the backwards propagation of error.
With the delta rule, as with other types of backpropagation, 'learning' is a supervised process that occurs with each cycle or 'epoch' (i.e. each time the network is presented with a new input pattern) through a forward activation flow of outputs, and the backwards error propagation of weight adjustments. More simply, when a neural network is initially presented with a pattern it makes a random 'guess' as to what it might be. It then sees how far its answer was from the actual one and makes an appropriate adjustment to its connection weights.The other learning rule that is mostly used is feed-forward type of neural network.