|
|
|
In the following section, we will define some of the terms used in the server. Some of the definitions may be vague. The readers are advised to view the contents under Parental Guidance (rated as PG).
|
SEQUENCE: I do not think this term needs any description. Except that, the sequence should be a protein. No carbohydrates or nucleic acids. The antigen sequence can be obtained from any database. NCBI (accessible via http://www.ncbi.nlm.nih.gov/) is one example. You can either paste it in the sequence box. Alternatively, you may submit a sequence file.
The server can accept both the formatted or unformatted(RAW) sequences. It uses ReadSeq routine to parse the input
-
FORMAT: This is obligatory. In addition, care should be taken that if you selected this option wrongly the results may not be of your expectations. Different databases use different sequence formats. However, you do not have to worry, just select the formatted option.
ALLELE SELECTION: The MHC molecules (particularly the class-I) are highly polymorphic. Polymorphic means many different variants. Each individual may have at the most six different alleles. Therefore, to predict the binding more practically the method should include maximum number of alleles. This is practically not feasible because some of the alleles are very little studied. The ProPred-I implements matrix-based prediction models for 47 class-I MHC alleles. The users can select single, multiple or all alleles.
RESULT: The ProPred-I offer four different result display modes. A brief description of these modes is as follows:
GRAPHICAL
The graphical output represents the quantitative estimation of the MHC binding propensity along the sequence. Each binder is represented as a peak crossing the dashed threshold line. Besides this, the server plots the threshold profile. Here the number of predicted binders at different thresholds is represented as bars. This profile assist experienced users in selecting the threshold for locating the promiscuous regions. The graphics is generated in GIF format using the GDlibrary of PERL.
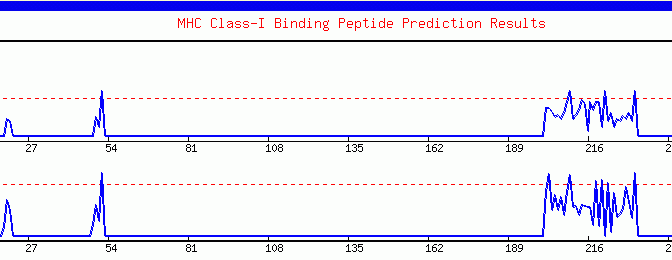
Graphical output generated by the ProPred-1. The peaks (starting from ~200th amino acid) crossing the red threshold line are the predicted binders.
TEXT
The text format is expressive in locating the promiscuous or allele specific binding regions. The text display has two options.
The first option (HTML-II) is similar to that used by TEPITOPE and ProPred. Here each binding regions are represented in blue with the first residue of each binding region in red color. Though useful, this option is less expressive in presenting the overlapping binding regions.

The HTML-II output: The peptide "KRGYNEDEV" is predicted to bind to HLA-B14, HLA-B*2702, and HLA-B*2705. On the other hand, the "QRIEELDHEL" is predicted to bind to five alleles (which one?). So you see the second peptide is more promiscuous
In second option (HTML-I), the overlapping regions are presented on separate lines making it easier to detect the overlaps.

The HTML-I output: The peptide "KRGYNEDEV" is predicted to bind to HLA-B*2702, and HLA-B*2705. On the other hand, the "LRQRIEELDHEL" is predicted to bind to two alleles in five different frames (which one?).
TABULAR
This is the most widely used display mode. Therefore no details. In brief, the peptide frames are sorted according to their score, and user specified number of frames is displayed in the form of a table.
THRESHOLD: The Threshold is a pre-defined numerical value used to make decision.
The threshold is defined as the 'percentage of best scoring natural peptides'(PSC). For example, a threshold of 1% would predict peptides in any given protein sequence that belongs to the 1% best scoring natural peptides.
The threshold values are derived as follows:
- Peptide score for all valid Peptide Frames in SWISPROT are calculated for any given matrix.
-
Peptide Frames are sorted based on their Peptide Scores, and the score values corresponding to the 1%, 2%, 3%, etc. best scoring peptides are determined.
Thus, the threshold correlates with the PSC value and is therefore an indicator for the likelihood that the predicted peptides are capable of binding to a given HLA molecule.
The % threshold parameter allows the user to select different stringency levels, in order to modulate the prediction results: a lower threshold corresponds to a high stringency prediction, i.e. to a lower rate of false positives and to a higher rate of false negatives. In contrast, a higher threshold value (low stringency) corresponds to a higher rate of false positives and a lower rate of false negatives. In short, from the same protein sequence input, a threshold setting of 1% will predict a lower number of peptide sequences and for a lower number of alleles, compared to 2% or higher thresholds; however, this will ensure a higher likelihood of positive downstream experimental results. Normally, at least for a first round of screening, threshold values higher than 3% are not desirable, since the rate of false positives can increase the size of the predicted repertoire to an amount unacceptable for later experimental testing.
PROTEASOME AND IMMUNOPROTEASOME FILTERS:
Evidences suggest that the cleavage specificities of proteasomes play an important role in presenting peptides to the class-I MHC molecules. Therefore, researchers tried to improve the MHC binding predictions with the proteasomal cleavage predictions. In ProPred-I, we implemented filters for the proteasomal and immunoproteasomal cleavages on the predicted MHC binders. Thus simulating two important steps of the antigen-processing pathway. The Proteasomes harboring the IFN-(-inducible subunits are known as immunoproteasomes. They process a number of viral epitopes with greater efficacy (Toes et al., 2001). A related server implementing both the MHC binding and proteasomal predictions, the MAPPP available at http://www.mpiib-berlin.mpg.de/MAPPP/.
|
|